MKSAP Quiz: Evaluation for follow-up of abdominal pain
A 35-year-old woman is evaluated for follow-up of abdominal pain, which has prompted multiple emergency department visits. CT scans have revealed intussusception. Her mother was diagnosed with breast cancer at age 40 years. The rest of her family history is unknown.
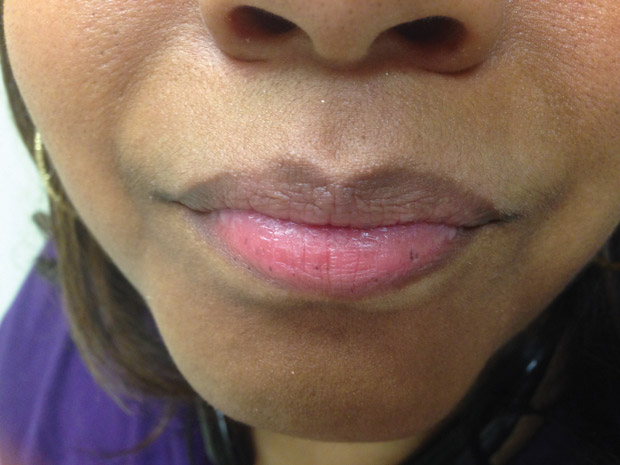
On physical examination, vital signs are normal. Findings on lips are shown.
Which of the following is the most likely diagnosis?
A. Adenomatous polyposis
B. Hamartoma syndrome (Cowden syndrome)
C. Juvenile polyposis syndrome
D. Peutz-Jeghers syndrome
Answer and critique
The correct answer is D. Peutz-Jeghers syndrome. This item is Question 71 in MKSAP 19's Gastroenterology and Hepatology section.
This patient likely has Peutz-Jeghers syndrome (PJS) (Option D), which is caused by mutations in the STK11 (LKB1) gene. PJS is a hamartomatous polyposis syndrome, and PJS-type polyps are found primarily in the small bowel as well as the stomach and colon. Small-bowel hamartomas develop at a young age (average age, 18.5 years), and patients may present with rectal bleeding (most commonly), bowel obstruction, and intussusception. A clinical diagnosis of PJS may be made when two of the following three criteria are met: two or more PJS-type hamartomatous polyps in the gastrointestinal tract; typical mucocutaneous hyperpigmentation (melanotic macules) in the mouth, buccal mucosa, nose, eyes, genitalia, or fingers; and family history of PJS. Patients with PJS have increased lifetime risk for multiple malignancies: colorectal (39%), gastric (29%), small bowel (13%), breast (32%-54%), ovarian/sex cord tumor with annular tubules (21%), cervical/adenoma malignum (1%), uterine (9%), testicular/Sertoli cell tumor (9%), lung (7%-17%), and pancreatic (11%-36%). The multiple melanotic macules present on this patient's lips are not seen in other familial polyposis syndromes. It is important to recognize that people without PJS may also have melanotic macules, most commonly on the lower lip, but macules may also be found on the gingiva, buccal mucosa, or tongue or within the genital mucosa. Solitary lesions are the rule, but multiple lesions suggest PJS.
Adenomatous polyposis syndromes (Option A) include familial adenomatous polyposis, which is caused by mutations in the APC gene, as well as MUTYH-associated polyposis, caused by bi-allelic loss of MUTYH. Mutations in polymerase proofreading genes POLE and POLD1 are rare but are also associated with adenomatous polyposis. These latter syndromes have features of both Lynch syndrome and familial adenomatous polyposis. The dermatologic findings observed in this patient are not seen in adenomatous polyposis syndromes.
Mutations in the tumor suppressor PTEN gene cause PTEN hamartoma syndrome (also known as Cowden syndrome) ( Option B) and are associated with a lifetime risk for colorectal cancer of 9% to 16%. Patients with this syndrome exhibit macrocephaly, facial trichilemmomas, lipomas, and oral papillomas as well as hamartomatous polyps.
Juvenile polyposis syndrome (Option C) is caused by mutations in SMAD4 and BMPR1A. Patients often present with rectal bleeding in their teens or early twenties. Evaluation can reveal multiple juvenile polyps (>5 polyps) in the colon (98%), stomach (14%), and small bowel (14%). It is important to differentiate juvenile polyposis syndrome from sporadic juvenile polyps, which may be seen in 1% of children and are not considered syndromic.
Key Point
- The diagnosis of Peutz-Jeghers syndrome (PJS) is based on the presence of two of the following three criteria: two or more PJS-type hamartomatous polyps in the gastrointestinal tract; multiple melanotic macules in the mouth, buccal mucosa, nose, eyes, genitalia, or fingers; and family history of PJS.


